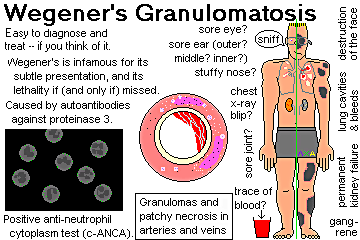
Wegener's granulomatosis
is an uncommon disease, characterized by inflammation of the blood vessels (vasculitis). This inflammation results in a reduction of oxygen in the blood and damages vital organs of the body by restricting blood flow to those organs and destroying normal tissue. Although vasculitis can damage any organ system, Wegener's granulomatosis primarily affects the respiratory tract (sinuses, nose, trachea [windpipe] and lungs) and the kidneys. This disorder can occur at any age and strikes men and women equally. It is rare in African Americans compared to whites. The cause of Wegener's granulomatosis is not known.
Symptoms
The initial symptoms of Wegener's granulomatosis are often vague or nonspecific and frequently include upper respiratory tract symptoms, joint pains, weakness, and fatigue.
Upper respiratory tract. The most common sign of Wegeners granulomatosis is involvement of the upper respiratory tract, which occurs in nearly all patients. Symptoms include sinus pain, discolored or bloody nasal drainage and, occasionally, nasal ulcerations. A common manifestation of the disease is persistent rhinorrhea ("runny nose") or other cold symptoms that do not respond to standard treatment or that become progressively worse. Rhinorrhea can result from nasal inflammation or sinus drainage and can cause pain. A hole or perforation of the nasal septum may develop, and collapse of the nasal bridge (called saddlenose deformity) may occur in some individuals. Blockage of the eustachian tubes, which are important for normal ear function, may cause chronic ear problems and hearing loss. A secondary bacterial infection can cause Wegeners-related sinusitis (inflammation of the sinuses) with congestion and chronic sinus pain.
Lungs. The lungs are affected in most patients with Wegeners granulomatosis, although no symptoms may be present. If symptoms are present, they include cough, hemoptysis (coughing up of blood), shortness of breath, and chest discomfort.
Kidneys. Kidney involvement, which occurs in more than three-fourths of patients, usually does not cause symptoms. If detected by blood tests, proper treatment can be started, preventing long-term damage to the kidneys.
Musculoskeletal system. Pain in the muscles and joints or, occasionally, joint swelling affects two-thirds of patients with Wegeners granulomatosis. Although joint pain can be very uncomfortable, it does not lead to permanent joint damage or deformities.
Eyes. Wegeners granulomatosis can affect the eyes in several ways. Patients may develop conjunctivitis (inflammation of the conjunctiva, the inner lining of the eyelid), scleritis (inflammation of the scleral layer, the white part of the eyeball), episcleritis (inflammation of the episcleral layer, the outer surface of the sclera), or as an orbital mass lesion (sore behind the eye globe). Uveitis or Intra-Ocular Inflammation is inflammation of the Uvea Tract, middle section of the eye. The symptoms of eye involvement include redness, burning or pain in the eye. Double vision or a decrease in vision are serious symptoms requiring immediate medical attention.
Skin lesions. Nearly half of people with Wegeners granulomatosis develop skin lesions. These small red or purple raised areas or blister-like lesions, ulcers, or nodules may or may not be painful.
Other symptoms. Some patients experience narrowing of the trachea (subglottic stenosis). The symptoms can include voice change, hoarseness, shortness of breath, or cough.
The nervous system and heart occasionally may be affected. Fever and night sweats also may occur. However, fever also may signal an underlying infection, often of the upper respiratory tract.
Diagnosis
To treat people with Wegeners granulomatosis most effectively, doctors must diagnose the disease early in its course. There are no blood tests that a doctor can use to diagnose Wegeners granulomatosis, but blood tests are important to rule out other causes of illness and to determine which organ sites may be affected. Most blood tests are nonspecific and can only suggest that a person has an inflammatory process. Anemia (low red blood cell count), elevated white blood cell count and platelet count, and an elevated sedimentation rate are commonly found in people with Wegeners granulomatosis. If the kidneys are involved, red blood cells and structures called red blood cell casts are visible in the urine when viewed under a microscope, and the blood tests measuring kidney function (creatinine and BUN) may show abnormalities.
X-ray results can be very helpful in diagnosing Wegeners granulomatosis. People with lung involvement will have abnormal chest x-rays, which may show one or many fluffy infiltrates, solid nodules, or cavities. Sinus x-rays or computed tomography (CT) scans in people with sinus involvement may show thickening of the sinus lining.
Many patients with active Wegener's granulomatosis have a blood test that reveals the presence of a specific type of antibody called antineutrophil cytoplasmic antibodies (ANCA) (an antibody is a disease-fighting protein). Although a positive ANCA test is useful in supporting a suspected diagnosis of Wegeners granulomatosis, in most instances it is not used by itself to make a diagnosis of this disorder. The ANCA test may be negative in some patients with active Wegeners granulomatosis.
Currently, the only definite way to diagnose Wegeners granulomatosis is by performing a biopsy of an involved organ site (usually the sinuses, lung, or kidney). The tissue is examined under the microscope to confirm the presence of vasculitis and granulomas (a specific type of inflammation), which together are diagnostic features of the disease. A biopsy is very important both to confirm the presence of Wegeners granulomatosis and also to assure the absence of other disorders that may have similar signs and symptoms.
Treatment
With the appropriate treatment, and diagnosed correctly and early, the outlook is good for patients with Wegeners granulomatosis.
In most cases, standard therapy consists of a combination of a glucocorticoid drug that reduces inflammation and a cytotoxic drug that interferes with the abnormal growth of cells.
Prednisone is the most common glucocorticoid drug (a steroid) that is used. Prednisone is similar to hydrocortisone, the natural glucocorticoid hormone produced by the body. It is chemically different from the anabolic steroids that have been used by athletes and is given in doses much higher than the body normally produces. Prednisone is usually administered as a single morning dose in an attempt to imitate how the body normally secretes hydrocortisone. When the persons illness improves, the prednisone dose is gradually decreased and converted to an every other day dosing schedule, usually over a period of 3 to 4 months. With further improvement in the disease, the prednisone is very gradually decreased and discontinued completely after approximately 6 to 12 months. When prednisone is taken by mouth, the body stops making its own natural hydrocortisone. As the prednisone dose is gradually reduced the body will resume making hydrocortisone again. It is extremely important that prednisone never be stopped suddenly because the body requires prednisone (or hydrocortisone) for its function and may not be able to immediately make what it needs
Cyclophosphamide (Cytoxanô) is the most commonly used cytotoxic drug. Cyclophosphamide is taken once a day by mouth. It is important for a patient to take the drug all at once in the morning followed by drinking a large amount of fluid. Although the initial dose of cyclophosphamide is based on the patients weight and kidney function, the doctor may adjust the dosage based on the blood counts, which are monitored closely to be sure that the white blood cell count is maintained at a safe level. Cyclophosphamide is continued for a full year beyond that point at which the disease has become quiet (is in remission). The dose of cyclophosphamide is then decreased gradually and eventually discontinued.
Cyclophosphamide and prednisone are both powerful drugs that suppress the immune system. Although these medications are beneficial in treating Wegeners granulomatosis, patients and their doctors should be aware that the drugs potentially have serious side effects. Careful monitoring by the doctor is very important. Because these drugs suppress the immune system, they can affect the bodys ability to fight off infection. Patients should report immediately any symptoms of infection and, specifically, any fever to their doctors. Prednisone can cause weight gain, cataracts, brittle bones, diabetes, and alterations in mood and personality. Cyclophosphamide can cause bone marrow suppression (lowering of blood counts), sterility, hemorrhagic cystitis (bleeding from the bladder) as well as other serious side effects.
Methotrexate, Azathioprine (also called Imuran).
Other medicines :
During the course of treating Wegener's granulomatosis, doctors often give their patients other medicines to prevent medicine-related side effects. These include
Trimethoprim/sulfamethoxazole (also called bactrim or septra) is given three times a week to prevent Pneumocystis carinii infection (a lung infection)
A medicine regimen is often given to prevent prednisone-related bone loss (osteoporosis)
Folic acid or folinic acid (also called leucovorin) are often given to people taking methotrexate
Approximately half of people with Wegeners granulomatosis may experience a return (relapse) of their disease. This occurs most frequently within two years of stopping medication, but potentially can occur at any point both during treatment or after stopping treatment. Thus, it is extremely important that patients continue to see their physicians regularly, both while they are on these medications, as well as after the medications have been stopped. Even while on medication, many patients are able to lead relatively normal lives and will remain in remission after therapy has been stopped completely.
NIAID and other parts of NIH support research on Wegener's granulomatosis and related forms of vasculitis at medical centers throughout the country through the extramural grants program. For example, NIH has recently awarded a $6.25 million, 5-year grant to establish the
Vasculitis Clinical Research Consortium (VCRC) .The multicenter VCRC will foster and facilitate clinical investigation in the inflammatory vasculitides, including Wegener's granulomatosis.
The VCRC will consist of four major U.S. vasculitis centers:
Boston University School of Medicine, Massachusetts
The Cleveland Clinic, Ohio
The Johns Hopkins Vasculitis Center, Baltimore, Maryland
The Mayo Clinic College of Medicine, Rochester, Minnesota